GENESIS Tutorial 7.2 (2022)
Domain Motion Enhanced (DoME) model for the domain closure of Ribose Binding Protein
This tutorial introduces how to setup input files with a coarse-grained model, Domain Motion Enhanced (DoME) model developed by Kobayashi et al. 1 and calculate MD simulations with the model in GENESIS. The DoME model is a valiant of residue base Go-type model by Karanicolas and Brooks 2 3 (hereafter KB Go-model.) The potential form is same with KB Go-model. The reference parameters of bond, angle, dihedral, native contact terms are taken from the native structure of a target protein. It is categorized as a single-basin model, where a single native structure will be the minimum of energy. However, the DoME model is developed for multi-domain proteins that have one more experimental structures. The model uses the Motion Tree that describes conformational changes in a protein developed by Koike et al. 4 The Motion Tree is a tree diagram calculated from two different structures of a protein and gives separation of domains and magnitude of rigid-body motion. The DoME model uses the separation and domain motion magnitude. The minimum depths of native interactions, \(ε_{ij}\), between i-th residue and j-th residue is determined by Motion Tree.
\[
\varepsilon_{ij} =
\begin{cases}
\frac{c}{M_{IJ}} & (i \in I,\ j \in J,\ I \ne J) \\
\frac{c}{M_{II}} = \frac{c}{M_{\text{criterion}}} & (i, j \in I)
\end{cases}
\]
where I and J are domains by separated Motion Tree. \(M_{IJ}\) is magnitude between domains I and J in Motion Tree. \(M_{criterion}\) is 5.0 angstrom where is the criteria used in the original paper. 4. A constant factor c is calibrated to keep a reasonable balance between the native contacts and the rest of the interactions.
Preparation
All the files required for this tutorial are hosted in the GENESIS tutorials repository on GitHub.
If you haven’t downloaded the files yet, open your terminal and run the following command (see more in Tutorial 1.1):
$ cd ~/GENESIS_Tutorials-2022
# if not yet
$ git clone https://github.com/genesis-release-r-ccs/genesis_tutorial_materials
If you already have the tutorial materials, let’s go to our working directory:
$ cd genesis_tutorial_materials/tutorial-7.2
This tutorial mainly explains how to create DoME and micro-mixing DoME models. There is two steps: (1) system setup and (2) production run.
$ ls
1_setup 2_production
1. Setup
To prepare input files, we need proceed the following five steps;
# Change directory for the system setup
$ cd 1_setup
$ ls
1-1_get_pdb 1-2_MMTSB 1-3_MotionTree 1-4_DoME_apo 1-5_DoME_ligand scripts
- Get PDB files for different states
- Genrate input files using KB-Go model from the MMTSB server
- Calculate Motion Tree from two structures of a single protein
- Generate parameter file of DoME model (for apo state) from 1 and 2
- Generate parameter file of DoME model (for apo state) with ligand effect
1.1 Get PDB files
In the 1st step, we can download PDB files with apo and holo states of RBD.
# Download PDB files (Apo and Holo)
$ 1-1_get_pdbs
$ wget http://www.rcsb.org/pdb/files/1urp.pdb
$ wget http://www.rcsb.org/pdb/files/2dri.pdb
$ vmd -dispdev text -e pdb_write_chain.tcl -args 1urp.pdb A
$ vmd -dispdev text -e pdb_write_chain.tcl -args 2dri.pdb A
$ ls
1urp.pdb 1urp_A.pdb 2dri.pdb 2dri_A.pdb
1.2 Get Go model files from the MMTSB server.
In the 2nd step, the input files (psf/pdb) are generated from the input files of KB-Go model. Then, the first procedure is same with that with the KB-Go model using the MMTSB server in tutorial 7.1. (Recently, generator of KB-Go model in MMTSB server is not available.)
# KB-Go model from MMTSB
$ ls
GO_1urp_A.Qdetails GO_1urp_A.pdb GO_2dri_A.Qdetails GO_2dri_A.pdb setup.tcl
GO_1urp_A.Qlist GO_1urp_A.seq GO_2dri_A.Qlist GO_2dri_A.seq
GO_1urp_A.param GO_1urp_A.top GO_2dri_A.param GO_2dri_A.top
# generate psf/pdb files for GO model
$ vmd -dispdev text -e setup.tcl
1.3 Domain analysis from Motion Tree
In the 3rd step, Program for Motion Tree is provided as a binary file from the site.
The program has several options to analysis of domain motions using two pdbs. However, to generate DoME model, please use default values (-tH 5.0, -tN 30). To see details in the method, please read the original paper. 4
# Motion Tree
$ cd ../1-3_MotionTree
# Copy Motion Tree Binary to working directory
$ cp ${DOWNLOAD}/mtntr_linux .
$ ./mtntr_linux ../1-1_get_pdbs/1urp_A.pdb A ../1-1_get_pdbs/2dri_A.pdb A
$ ls
MT.info MT.ps MT000.pdb MT000.ras MT001.pdb MT001.ras mtntr_linux
MT.ps shows Motion Tree between 1urp and 2dri. The tree map indicate these residues are divided into two domains.
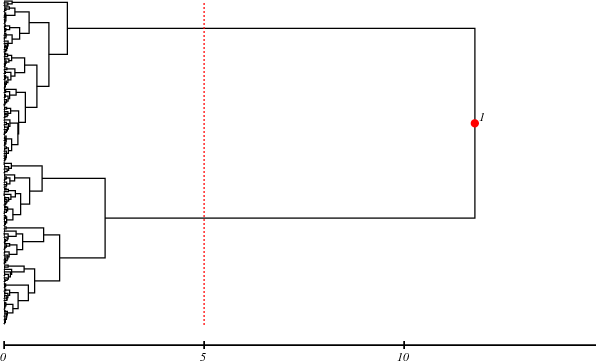
MT.info is information of domain separation. DoME model uses the information. Height indicates amplitude of domain motion. The residues (1-103, 233-265) are a domain (NTD), and (104-232, 266-271) are another domain (CTD):
$ cat MT.info
# Effective node: 1
@@ Height: 11.8
@@ Depth: 0
== Input1 ( Chain-ID: A ) ==
LL11: A larger part: 136 residues
LL12: Region: 1 - 103 , 233 - 265
SS11: A smaller part: 135 residues
SS12: Region: 104 - 232 , 266 - 271
== Input2 ( Chain-ID: A ) ==
LL21: A larger part: 136 residues
LL22: Region: 1 - 103 , 233 - 265
SS21: A smaller part: 135 residues
SS22: Region: 104 - 232 , 266 - 271
//
1.4 DoME model for a single state (apo state)
The perl script generating the DoME model is provided.
# DoME model of apo state
$ cd ../1-4_DoME_apo
# Perl script generating DoME model from KBGO and Motion Tree information.
# -param : parameter file of KBGO model. [in]
# -MT : Motion Tree information [in]
# -out : parameter file of DoME model. [out]
$ ../scripts/DoME.pl -param ../1-2_MMTSB/GO_1urp_A.param -MT ../1-3_MotionTree/MT.info -out DoME_single.param
Height: 11.8
$ ls
DoME_single.param
The strength of native contacts is shown (\(1.2>\varepsilon_{ij}>0.7\) : orange, \(\varepsilon_{ij}<0.7\) : blue)
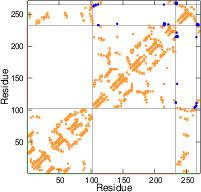
Interactions inside the domains are strong, while the interactions between the different domains are weaker.
Note: An input parameter of DoME model is constant, c in the above
equation. In this case, we use 1.158474 for the protein which is a
value of the strongest interaction of GO_2dri_A.param. (=holo state) You
want to use it to get a good balance with the holo state. Of course, you
can choose the value in the apo state (GO_1urp_A.param) if you just want
to compare KBGO model with the apo form. (You can change the constant by
using -const XXX in the script.)
1.5 DoME model with micro-mixing
The DoME model can be used a ligand binding model with micro-mixing perturbation. 5
# DoME model with micro-mixing state.
$ cd ../1-5_DoME_ligand
# Preparation of holo/apo parameters
$ ln -sf ../1-2_MMTSB/GO_1urp_A.param GO_apo.param
$ ln -sf ../1-2_MMTSB/GO_2dri_A.param GO_holo.param
$ ln -sf ../1-1_get_pdb/1urp_A.pdb apo.pdb
$ ln -sf ../1-1_get_pdb/2dri_A.pdb holo.pdb
# Generate interaction for micro-mixing perturbation term
$ ../scripts/ligand_cont.pl |& tee ligand_data
By comparing native contacts in holo and apo parameter files, the script
generates a new parameter file (GO.interdom.c1.200000.param) with
contacts applying perturbations. In the model, an interaction in
different domains showing large distance difference is considered as a
ligand interaction.
# Perl script generating DoME model from intermediate parameter file and Motion Tree information.
# -param : intermediate parameter file with ligands. [in]
# -MT : Motion Tree information [in]
# -fact : constants for ligand interactions
# -out : parameter file of DoME model. [out]
$ ../scripts/DoME_lig.pl -mt ../1-3_MotionTree/MT.info -param GO.interdom.c1.200000.param -out GO_apo_scaled_e1.7.param -fact 1.7
Height: 11.8
The strength of native contacts is shown (\(\varepsilon_{ij}>1.2\) : red, \(1.2>\varepsilon_{ij}>0.7\) : orange, \(\varepsilon_{ij}<0.7\) : blue) The red one indicates perturbated ligand contacts.
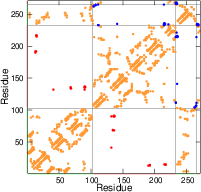
2. Production
# MD simulations
$ cd ../../2_production
Inut parameters of MD simulations are almost same with those in KB-Go model except for the parfile.
[INPUT]
topfile = ../1_setup/1-2_MMTSB/GO_1urp_A.top # topology file (apo state)
parfile = ../1_setup/1-4_DoME_apo/DoME_single.param # parameter file
psffile = ../1_setup/1-2_MMTSB/go_1urp.psf # protein structure file (apo state)
pdbfile = ../1_setup/1-2_MMTSB/go_1urp.pdb # reference for restraints (apo state)
For the micro-mixing model, the DoME with ligand paramter is used while the other files (psf/top) are paramter files with apo state.
[INPUT]
topfile = ../1_setup/1-2_MMTSB/GO_1urp_A.top # topology file (apo state)
parfile = ../1_setup/1-5_DoME_ligand/GO_apo_scaled_e1.7.param # parameter file
psffile = ../1_setup/1-2_MMTSB/go_1urp.psf # protein structure file (apo state)
pdbfile = ../1_setup/1-2_MMTSB/go_1urp.pdb # reference for restraints (apo state)
Written by Chigusa Kobayashi@RIKEN Center for Computational Science, April 2022