GENESIS Tutorial 12.1 (2022)
REMD simulation of (Ala)3 in water
In this tutorial, we illustrate how to run temperature replica exchange molecular dynamics (T-REMD) simulations of (Ala)3 and calculate PMF of end to end distance and dihedral angle (Φ and Ψ) distribution. T-REMD is an enhanced sampling method that parallelly simulate the system at a range of different temperatures and periodically exchanging between them. As a result of sampling at several temperature, the simulation can efficiently overcome well known problem of conventional MD, wherein the conformation could be trapped in a local minimum. For further details of REMD method, please refer to the original paper.1
REMD simulation in GENESIS requires an MPI environment.
At least one MPI processor must be assigned to one replica.
Please note that it may take more than 12 hours
to finish all simulations in this tutorial.
python 3.x.x is required for analysis in this tutorial.
Preparation
All the files required for this tutorial are hosted in the GENESIS tutorials repository on GitHub. If you haven’t downloaded the files yet, open your terminal and run the following command (see more in Tutorial 1.1):
$ cd ~/GENESIS_Tutorials-2022
# if not yet
$ git clone https://github.com/genesis-release-r-ccs/genesis_tutorial_materials
If you already have the tutorial materials, let’s go to our working directory:
$ cd genesis_tutorial_materials/tutorial-12.1
This tutorial consists of five steps: 1) system setup, 2) energy minimization and pre-equilibration, 3) REMD equilibration, 4) REMD production, and 5) trajectory analysis. Control files for GENESIS are already included in the download file. Since we use the CHARMM36m force field parameters,2 we make a symbolic link to the CHARMM toppar directory (see Tutorial 2.2).
$ ln -s ../../Data/Parameters/toppar_c36_jul21 ./toppar
$ ls
1_setup 2_min_equil 3_equil_remd 4_prod_remd 5_analy toppar
1. Setup
In this tutorial, we simulate (Ala)3 in explicit water. We use the same initial structure as in Tutorial 3.2.
# Change directory for the system setup
$ cd 1_setup
# Prepare PDB and PSF files
$ ln -s ../../tutorial-3.2/1_setup/3_solvate/wbox.pdb ./
$ ln -s ../../tutorial-3.2/1_setup/3_solvate/wbox.psf ./
2. Minimization and pre-equilibration
We use the same protocols described in Tutorial 3.2. Since we have already equilibrated the system in Tutorial 3.2, we use the restart file obtained previously.
# Change directory for the minimization and equilibration
$ cd ../2_minimize_pre-equi
# Prepare equilibrated restart file
$ ln -s ../../tutorial-3.2/3_equilibrate/eq3.rst
3. REMD equilibration
Temperature setting
Before starting T-REMD simulation, we must determine the number of replicas and the temperatures of replicas. These temperatures need to be chosen carefully, because they will greatly affect the results of T-REMD simulations and probabilities of the replica exchanges. If the difference of temperatures between adjacent replicas is small, the exchange probability is high, but the number of replicas needed for such simulation is also large. Therefore, we need to find the optimal temperature intervals to perform efficient REMD simulations.
We recommend using the web server REMD Temperature generator.3 This tool automatically generates the number of replicas and their temperatures according to the information we input. We show the example of the input for the T-REMD simulation of the solvated trialanine system:
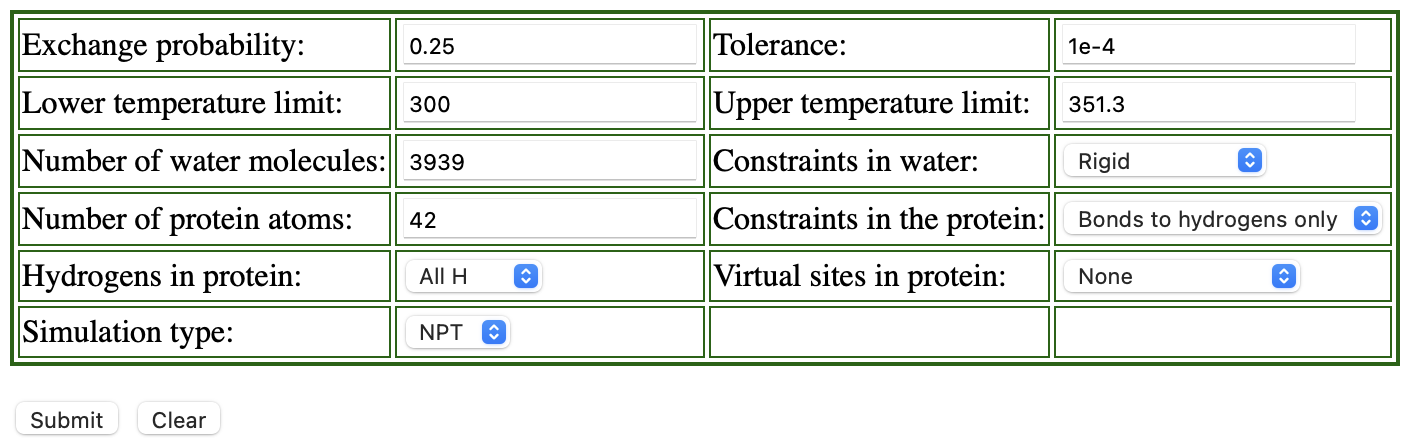
Replica exchange probabilities are often set to 0.2 – 0.3, and here we set the replica exchange probability to 0.25.
The lower and upper temperature limits are set to 300 K and 351.3 K, respectively.
Since we use SHAKE and SETTLE algorithms in the simulation,
“constraints in the protein” is set to “Bonds to hydrogen only”,
and “Constraints in water” to “rigid”.
We use all-atom model, so we select “All H” for the parameter of “Hydrogens in proteins”.
The numbers of protein atoms and the water molecules are input according to the numbers in ../1_setup/wbox.pdb.
We can count the number of the water molecules in the system as follows:
# Count the number of TIP3P water molecules
$ grep "TIP3" ../1_setup/wbox.pdb | wc -l | awk '{print $1 / 3}'
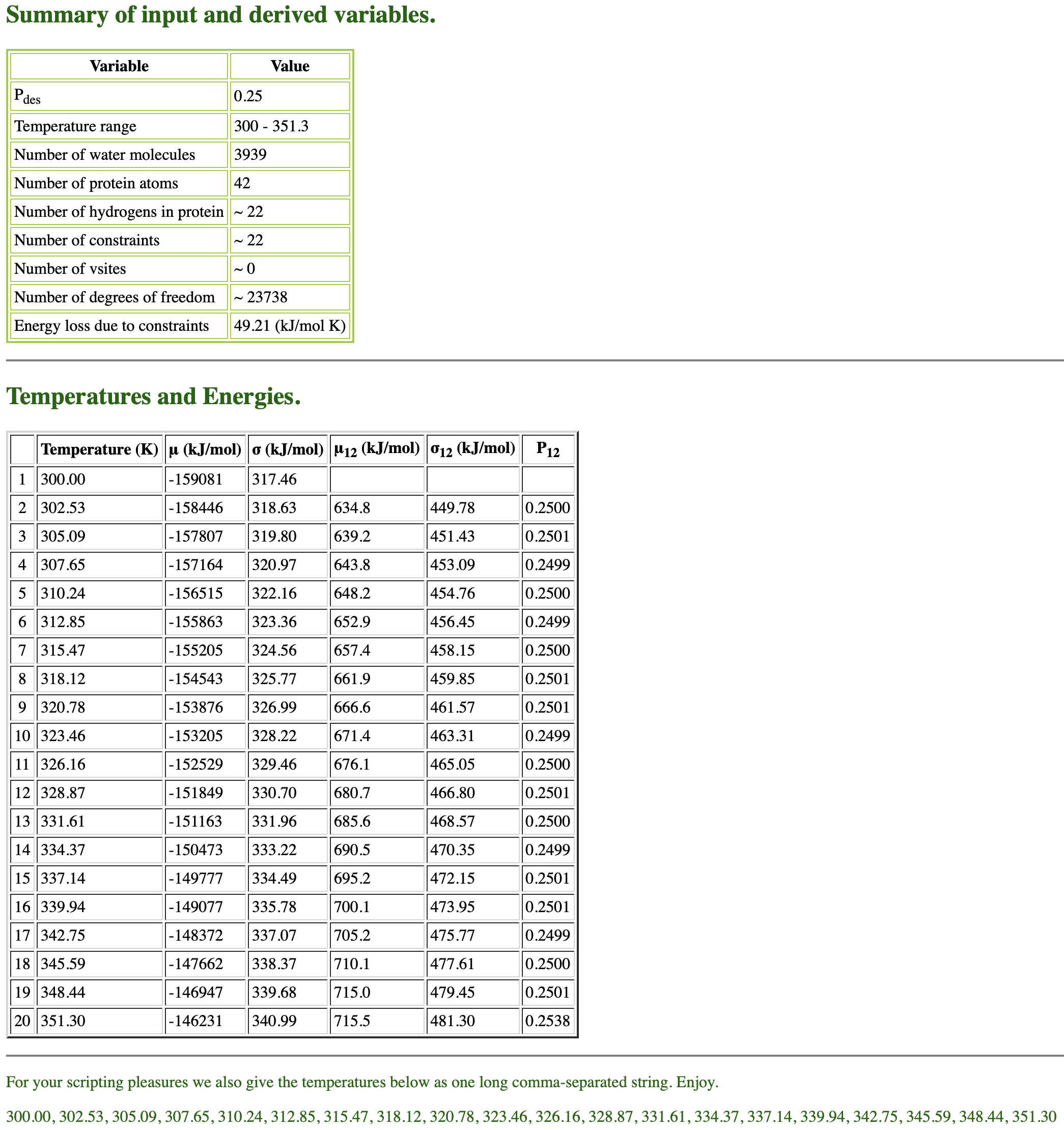
When we fill in all the parameters and submit them, the server generates
the summary, temperatures and energies of 20 replicas in a few seconds
as follows.
In the table shown above, there is the parameter “Simulation type”. We
can select either “NPT” or “NVT” for the parameter, but, when we select
“NVT”, the server returns an error message. Thus we choose “NPT” here,
even though we use NVT ensemble in our T-REMD simulation.
Equilibration
# Change directory for the REMD equilibration
$ cd ../3_equil_remd
$ ls
run.inp
Using generated temperature parameters, we start our REMD simulation.
First, each replica must be equilibrated at the selected temperature
just like conventional MD simulations. The following command performs a
500 ps NVT MD simulations with run.inp. This equation of motion are
integrated with the RESPA integrator (VRES) with the time step of 2.5
fs, where the SHAKE algorithm is used for bond constraint. Make sure
that you use NVT ensemble, as using NPT ensemble make significant
differences in the potential energy of each replica and subsequently
hinder the exchange between them. The REMD equilibration control file
is:
[INPUT]
topfile = ../toppar/top_all36_prot.rtf # topology file
parfile = ../toppar/par_all36m_prot.prm # parameter file
strfile = ../toppar/toppar_water_ions.str # stream file
psffile = ../1_setup/wbox.psf # protein structure file
pdbfile = ../1_setup/wbox.pdb # PDB file
reffile = ../1_setup/wbox.pdb # PDB file
rstfile = ../2_min_equil/eq3.rst # restart file
[OUTPUT]
dcdfile = run_rep{}.dcd # DCD trajectory file
rstfile = run_rep{}.rst # restart file
remfile = run_rep{}.rem # replica exchange ID file
logfile = run_rep{}.log # log file of each replica
[ENERGY]
forcefield = CHARMM # CHARMM force field
electrostatic = PME # use Particle mesh Ewald method
switchdist = 10.0 # switch distance
cutoffdist = 12.0 # cutoff distance
pairlistdist = 13.5 # pair-list distance
pme_nspline = 4 # order of the spline interpolation
vdw_force_switch = YES # turn on vdw force switch
pme_max_spacing = 1.2 # Max grid spacing allowed
[DYNAMICS]
integrator = VRES # [LEAP,VVER,VRES]
nsteps = 200000 # number of MD steps
timestep = 0.0025 # timestep (ps)
eneout_period = 2000 # energy output period
crdout_period = 2000 # coordinates output period
rstout_period = 20000 # restart output period
nbupdate_period = 10 # pairlist update period
elec_long_period = 2 # period of reciprocal space calculation
thermostat_period = 10 # period of thermostat update
barostat_period = 10 # period of barostat update
[CONSTRAINTS]
rigid_bond = YES # use SHAKE/RATTLE
fast_water = YES # use SETTLE
[ENSEMBLE]
ensemble = NVT # [NVE,NVT,NPT]
tpcontrol = BUSSI # thermostat and barostat
temperature = 300 # K
group_tp = YES # group temperature/pressure evaluation
[BOUNDARY]
type = PBC # periodic boundary condition
[REMD]
dimension = 1 # number of parameter types
exchange_period = 0 # NO exchange for equilibration
type1 = TEMPERATURE # T-REMD
nreplica1 = 20 # number of replicas
parameters1 = 300.00 302.53 305.09 307.65 310.24 \
312.85 315.47 318.12 320.78 323.46 \
326.16 328.87 331.61 334.37 337.14 \
339.94 342.75 345.59 348.44 351.30
In [INPUT] section, we specify ../2_min_equil/eq3.rst file as the
restart file of the simulation run. In the REMD simulation, the 20
(equal to the number of replicas specified nreplica in [REMD] section explained below) copies are automatically made from this restart
file.
In [OUTPUT] section, in addition to dcdfile and rstfile names, we give
also logfile and remfile. If we perform REMD simulations, file names of
logfile and remfile are also required. logfile gives a log file of MD
simulation for each replica. The remfile gives replica exchange
parameter. In this section, “{}” returns a replica index number from 1
to 20.
When we want to run REMD simulations, we add [REMD] section in the
control file. In [REMD] section, the number of dimensions is set
to dimension = 1, and the type of the exchanged variable is set
to type1 = temperature for T-REMD simulation. The number of replicas
is set to nreplica1 = 20, and we assign the replica temperatures to
the variable parameter1. If exchange_period = 0, then no exchanges
occur during the run, and so we set exchange_period to 0 for
equilibration of all replicas.
In [ENSEMBLE] section, we use NVT ensemble in this tutorial. Though
we can perform REMD simulations both in NVT and NPT ensemble with
GENESIS, we recommend performing T-REMD simulations in NVT ensemble,
because in a temperature exceeding the water boiling point, the system
may be disrupted.
To run REMD equilibiration, we use the following commands. Here we used 8 MPI processes and 4 OpenMP threads for each replica, i.e., a total of 640 (= 8 x 4 x 20) CPU cores.
# Run REMD-equilibiration step.
$ export OMP_NUM_THREADS=4
$ mpirun -np 160 /home/user/GENESIS/bin/spdyn run.inp > run.log
4. REMD production
# Change directory for the REMD production
$ cd ../4_prod_remd
$ ls
run.inp
Since we have now completed all preparation steps, now we can start running the production simulation. We ran a short simulation for 10 ns for the purpose of tutorial, however longer simulation is actually needed to obtain better energy results. The following control file is used to run the simulation in NVT ensemble:
[INPUT]
topfile = ../toppar/top_all36_prot.rtf # topology file
parfile = ../toppar/par_all36m_prot.prm # parameter file
strfile = ../toppar/toppar_water_ions.str # stream file
psffile = ../1_setup/wbox.psf # protein structure file
pdbfile = ../1_setup/wbox.pdb # PDB file
reffile = ../1_setup/wbox.pdb # PDB file
rstfile = ../3_equil_remd/run_rep{}.rst # restart file
[OUTPUT]
dcdfile = run_rep{}.dcd # DCD trajectory file
rstfile = run_rep{}.rst # restart file
remfile = run_rep{}.rem # replica exchange ID file
logfile = run_rep{}.log # log file of each replica
[ENERGY]
forcefield = CHARMM # CHARMM force field
electrostatic = PME # use Particle mesh Ewald method
switchdist = 10.0 # switch distance
cutoffdist = 12.0 # cutoff distance
pairlistdist = 13.5 # pair-list distance
pme_nspline = 4 # order of the spline interpolation
vdw_force_switch = YES # turn on vdw force switch
pme_max_spacing = 1.2 # Max grid spacing allowed
[DYNAMICS]
integrator = VRES # [LEAP,VVER,VRES]
nsteps = 4000000 # number of MD steps
timestep = 0.0025 # timestep (ps)
eneout_period = 2000 # energy output period
crdout_period = 2000 # coordinates output period
rstout_period = 20000 # restart output period
nbupdate_period = 10 # pairlist update period
elec_long_period = 2 # period of reciprocal space calculation
thermostat_period = 10 # period of thermostat update
barostat_period = 10 # period of barostat update
[CONSTRAINTS]
rigid_bond = YES # use SHAKE/RATTLE
fast_water = YES # use SETTLE
[ENSEMBLE]
ensemble = NVT # [NVE,NVT,NPT]
tpcontrol = BUSSI # thermostat and barostat
pressure = 1 # atm
temperature = 300 # K
group_tp = YES # group temperature/pressure evaluation
[BOUNDARY]
type = PBC # periodic boundary condition
[REMD]
dimension = 1 # number of parameter types
exchange_period = 2000 # NO exchange for equilibration
type1 = TEMPERATURE # T-REMD
nreplica1 = 20 # number of replicas
parameters1 = 300.00 302.53 305.09 307.65 310.24 \
312.85 315.47 318.12 320.78 323.46 \
326.16 328.87 331.61 334.37 337.14 \
339.94 342.75 345.59 348.44 351.30
In [INPUT] section, rstfile points to the output files from the
equilibration. “{}” returns a series of the 20 input files.
In [REMD] section, the period between replica exchanges is
specified by exchange_period = 2000. Then replica exchange attempts
occurs every 2000 steps, that is, every 5 (=2000 x 0.025) ps.
The other parameters in [REMD] section should not be changed from
those in the input files of your previous runs.
To run REMD production, we use the following commands. Here we used 8 MPI processes and 4 OpenMP threads for each replica, i.e., a total of 640 (= 8 x 4 x 20) CPU cores.
# Run REMD production
$ export OMP_NUM_THREADS=4
$ mpirun -np 160 /home/user/GENESIS/bin/spdyn run.inp > run.log
5. Analysis
In this tutorial, we mainly focus on calculating PMF of the end to end distance and dihedral angle distribution at 300 K. In which, we use all temperatures trajectory upon applying the Multistate Bennett Acceptance Ratio (MBAR) re-weighting method. For information on MBAR method, please refer to the original paper.4
End to end distance of (Ala)3
Dihedral angle of (Ala)3
In REMD control file, we setup the exchange_period=2000. In the log
output of the REMD simulation, we can see the information about
replica-exchange attempts at every exchange_period steps.
REMD> Step: 1998000 Dimension: 1 ExchangePattern: 2
Replica ExchangeTrial AcceptanceRatio Before After
1 3 > 2 R 148 / 500 305.090 305.090
2 6 > 7 A 171 / 500 312.850 315.470
3 1 > 0 N 0 / 0 300.000 300.000
4 4 > 5 R 170 / 500 307.650 307.650
5 11 > 10 R 199 / 500 326.160 326.160
6 7 > 6 A 171 / 500 315.470 312.850
7 8 > 9 R 178 / 500 318.120 318.120
8 17 > 16 A 198 / 500 342.750 339.940
9 2 > 3 R 148 / 500 302.530 302.530
10 20 > 0 N 0 / 0 351.300 351.300
11 18 > 19 A 200 / 500 345.590 348.440
12 13 > 12 R 208 / 500 331.610 331.610
13 10 > 11 R 199 / 500 323.460 323.460
14 9 > 8 R 178 / 500 320.780 320.780
15 5 > 4 R 170 / 500 310.240 310.240
16 12 > 13 R 208 / 500 328.870 328.870
17 15 > 14 R 184 / 500 337.140 337.140
18 16 > 17 A 198 / 500 339.940 342.750
19 14 > 15 R 184 / 500 334.370 334.370
20 19 > 18 A 200 / 500 348.440 345.590
Parameter : 305.090 315.470 300.000 307.650 326.160 312.850 318.120 339.940 302.530 351.300 348.440 331.610 323.460 320.780 310.240 328.870 337.140 342.750 334.370 345.590
RepIDtoParmID: 3 7 1 4 11 6 8 16 2 20 19 13 10 9 5 12 15 17 14 18
ParmIDtoRepID: 3 9 1 4 15 6 2 7 14 13 5 16 12 19 17 8 18 20 11 10
REMD> Step: 2000000 Dimension: 1 ExchangePattern: 1
Replica ExchangeTrial AcceptanceRatio Before After
1 3 > 4 A 183 / 500 305.090 307.650
2 7 > 8 R 173 / 500 315.470 315.470
3 1 > 2 R 240 / 500 300.000 300.000
4 4 > 3 A 183 / 500 307.650 305.090
5 11 > 12 R 152 / 500 326.160 326.160
6 6 > 5 A 167 / 500 312.850 310.240
7 8 > 7 R 173 / 500 318.120 318.120
8 16 > 15 A 174 / 500 339.940 337.140
9 2 > 1 R 240 / 500 302.530 302.530
10 20 > 19 A 229 / 500 351.300 348.440
11 19 > 20 A 229 / 500 348.440 351.300
12 13 > 14 A 177 / 500 331.610 334.370
13 10 > 9 R 153 / 500 323.460 323.460
14 9 > 10 R 153 / 500 320.780 320.780
15 5 > 6 A 167 / 500 310.240 312.850
16 12 > 11 R 152 / 500 328.870 328.870
17 15 > 16 A 174 / 500 337.140 339.940
18 17 > 18 R 177 / 500 342.750 342.750
19 14 > 13 A 177 / 500 334.370 331.610
20 18 > 17 R 177 / 500 345.590 345.590
Parameter : 307.650 315.470 300.000 305.090 326.160 310.240 318.120 337.140 302.530 348.440 351.300 334.370 323.460 320.780 312.850 328.870 339.940 342.750 331.610 345.590
RepIDtoParmID: 4 7 1 3 11 5 8 15 2 19 20 14 10 9 6 12 16 17 13 18
ParmIDtoRepID: 3 9 4 1 6 15 2 7 14 13 5 16 19 12 8 17 18 20 10 11
In this log file, we should pay attention to the AcceptanceRatio
values. In this table, 'A' and 'R' mean that the exchange at this
step is accepted or rejected respectively. The last two columns show
replica temperatures before and after the exchange trials respectively.
The Parameter, RepIDtoParmID, and ParmIDtoRepID lines summarize the locations and parameters after replica exchanges.
The Parameter line gives the temperature of each replica in T-REMD simulation.
The RepIDtoParmID line stands for the permutation function that converts Replica ID to Parameter ID.
For example, in the 1st column, 4 is written, which means that the temperature of Replica 1 is set to 307.65 K.
The ParmIDtoRepID line also represents the permutation function that converts Parameter ID to Replica ID.
For example, in the 5th column, 6 is written, which means that Parameter 5 (corresponding to the replica temperature, 310.24 K) is located in Replica 6.
In REMD preparation step using temperature generator, we setup the probability of exchange to 0.25. So we first check the simulation acceptance ratio and check if it matches our original selection, see next section.
# change directory
$ cd ../5_analy
$ ls
1_calc_ratio 3_sort 5_end_end_distance 7_MBAR 9_PMF_dihedral
2_plot_index 4_plot_potential 6_dihedral_angle 8_PMF_distance
5.1. Calculate the acceptance ratio of each replica
As we mentioned in the previous section, acceptance ratio of replica
exchange is one of the important factors that determine the efficiency
of REMD simulations. The acceptance ratio is displayed in a standard log
output “../4_prod_remd/run.log”, and we examine the data from the last
step. Here, we show an example how to examine the data. Note that the
acceptance ratio of replica “A” to “B” is identical to “B” to “A”, and
thus we calculate only “A” to “B”. For this calculation, you can use the
script “calc_ratio.sh”.
# change directory
$ cd 1_calc_ratio
# make the file executable and use it
$ chmod u+x calc_ratio.sh
$ ./calc_ratio.sh
1 > 2 0.399
3 > 4 0.401
5 > 6 0.379
7 > 8 0.369
9 > 10 0.415
11 > 12 0.393
13 > 14 0.414
15 > 16 0.425
17 > 18 0.434
19 > 20 0.413
The file “calc_ratio.sh” contains the following commands:
# get acceptance ratios between adjacent parameter IDs
$ grep " 1 > 2" ../../4_prod_remd/run.log | tail -1 > acceptance_ratio.dat
...
...
$ grep " 19 > 20" ../../4_prod_remd/run.log | tail -1 >> acceptance_ratio.dat
# calculate the ratios
awk '{printf ("%2d %s %2d %4.3f \n", $2,$3,$4,$6/$8)}' acceptance_ratio.dat
5.2. Plot time courses of replica indices and temperatures
To examine the random walks of each replica in temperature space, we
analyze “time course of the replica indices for each set temperature”
and “time courses of temperatures in one replica”. First, we analyze
time course of the replica indices for each set temperature. We need to
plot the values of the “ParmIDtoRepID” lines from
../4_prod_remd/run.log for a chosen starting replica temperature
versus time. Each column correspond to initial set temperature
respectively. For example, the first column correspond to 300 K, the 10th column correspond to 323.46 K, and the last column correspond to 351.3 K. Using following commands, we can get
replica IDs in each snapshot.
# change directory
$ cd ../2_plot_index
$ ls
$ plot_index.sh plot_temperature.sh plot_repID-Tempreture.gnuplot plot_parmID-repID.gnuplot
# make the file executable and use it
$ chmod u+x plot_index.sh
$ ./plot_index.sh
# plot replica IDs in each snapshot
$ gnuplot plot_parmID-repID.gnuplot
The file “plot_index.sh” contains the following commands:
# get replica IDs in each snapshot
grep "ParmIDtoRepID:" ../../4_prod_remd/run.log | sed 's/ParmIDtoRepID:/ /' > T-REMD_parmID-repID.dat
The file “plot_parID-repID.gnuplot” contains the following commands :
set terminal jpeg
reset
set yrange [0:22]set mxtics
set mytics
set xlabel "Time (ns)"
set ylabel "Replica ID"
set output "300.00k.jpg"
plot "T-REMD_parmID-repID.log" using ($1*0.0025*0.001):2 with points pt 5 ps 0.5 lt 1 title "300.00 K"
set output "323.46k.jpg"
plot "T-REMD_parmID-repID.log" using ($1*0.0025*0.001):11 with points pt 7 ps 0.5 lt 2 title "323.46 K"
set output "351.30k.jpg"
plot "T-REMD_parmID-repID.log" using ($1*0.0025*0.001):21 with points pt 9 ps 0.5 lt 3 title "351.30 K"
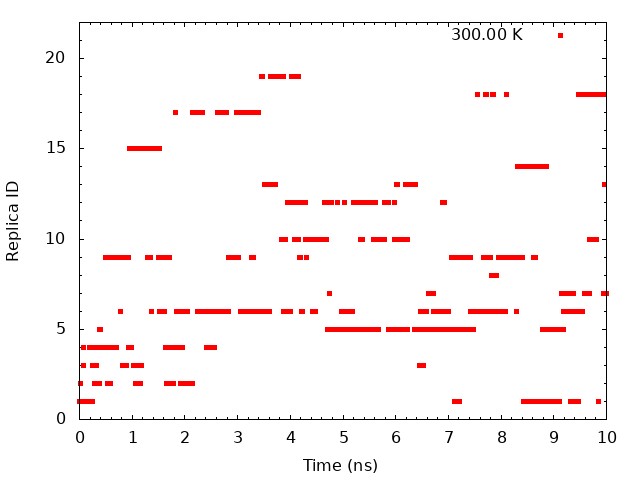
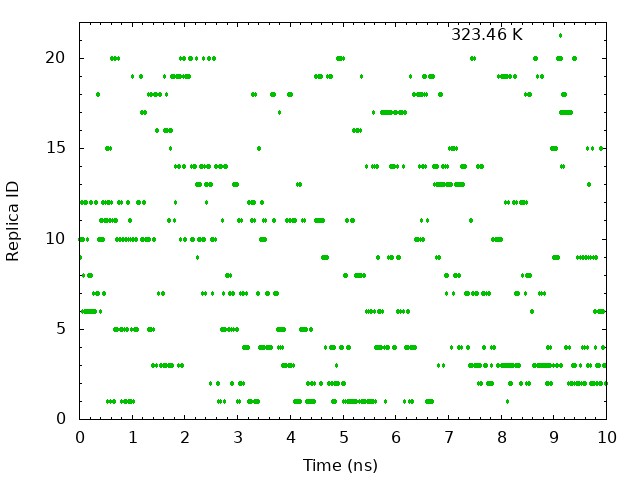
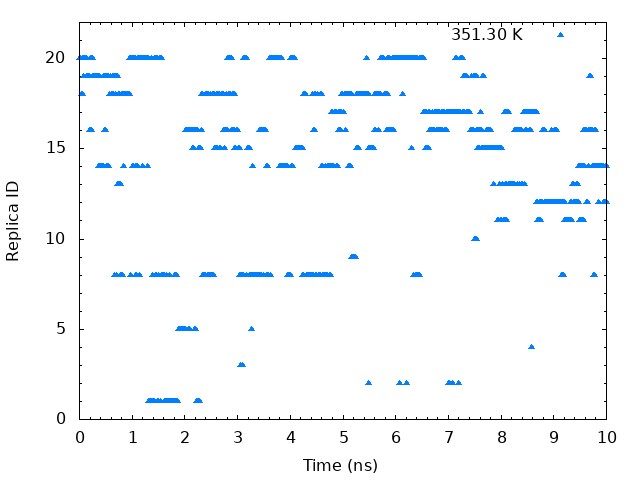
These graphs indicate that a certain temperature (parameterID) visit randomly each replica, and thus random walks in the temperature spaces are realized.
Next, we plot time courses of temperatures in one replica. We need to
plot one column in the “Parameter :” lines in ../4_prod_remd/run.log
versus time. Using following commands. we can get replica temperatures
in each snapshot.
# make the file executable and use it
$ chmod u+x plot_temperature.sh
$ ./plot_temperature.sh
# plot tempreture IDs in each snapshot
$ gnuplot plot_repID-Temperature.gnuplot
The file “plot_temperature.sh” contains the following commands:
# get replica temperatures in each snapshot
grep "Parameter :" ../../4_prod_remd/run.log | sed 's/Parameter :/ /' > T-REMD_repID-temperatrue.dat
The “plot_repID-Temperature.gnuplot” include the following commands:
set terminal jpeg
set output "RepID1_10_20.jpg"
set yrange [300:360]set mxtics
set mytics
set xlabel "Time (ns)"
set ylabel "Temperature (K)"
plot \
"T-REMD_repID-Temperature.log" using ($1*0.0025*0.001):2 with lines lt 3 title "repID=1 ",\
"T-REMD_repID-Temperature.log" using ($1*0.0025*0.001):11 with lines lt 2 title "repID=10",\
"T-REMD_repID-Temperature.log" using ($1*0.0025*0.001):21 with lines lt 1 title "repID=20"
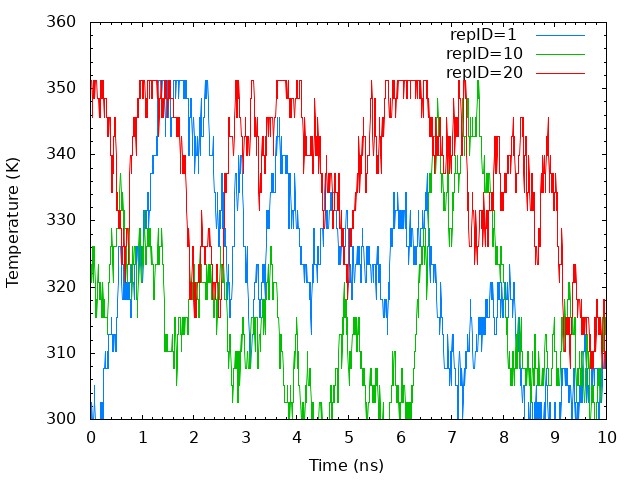
The temperatures of each replica during the simulation are distributed in all temperatures assigned. It means that correct annealing of the system is realized.
5.3. Sort coordinates in DCD trajectory files by parameters
The temperature in output DCD files of T-REMD simulation have all range
of temperatures, due to the exchange. Therefore, to analyze the
simulation further, we first need to sort the frames in the trajectory
based on their temperature. To do that, we use GENESIS analysis tool
remd_convert. Sorting is done based on the information written in
remfiles generated from the REMD simulation. Concomitantly, we also sort
log files for each replica based on temperature parameters
# change directory
$ cd ../3_sort
$ ls
$ remd_convert.inp
# Sort frames by parameters
$ /home/user/GENESIS/bin/remd_convert remd_convert.inp | tee remd_convert.log
The following control file is used to convert dcd files:
[INPUT]
psffile = ../../1_setup/wbox.psf
reffile = ../../1_setup/wbox.pdb
dcdfile = ../../4_prod_remd/run_rep{}.dcd
remfile = ../../4_prod_remd/run_rep{}.rem
logfile = ../../4_prod_remd/run_rep{}.log
[OUTPUT]
pdbfile = select.pdb
trjfile = parmID{}.dcd
logfile = parmID{}.log
[SELECTION]
group1 = segid:PROA
group2 = resno:2 and (an:N or an:CA or an:C or an:O)
[FITTING]
fitting_method = TR+ROT
fitting_atom = 2
mass_weight = YES
[OPTION]
check_only = NO
convert_type = PARAMETER
convert_ids = # (empty = all)
num_replicas = 20
nsteps = 4000000
exchange_period = 2000
eneout_perio = 2000
crdout_period = 2000
trjout_format = DCD
trjout_type = COOR+BOX
trjout_atom = 1
pbc_correct = NO
Now we have sorted temperature log and DCD file which will be used in the following analysis steps.
5.4. Plot potential energy distribution for each temperature
Now as we already sorted the log files for each temperature parameters, we plot potential energy distribution to ensure sufficient overlap between all parameters. First, grep command we extract potential energies and step number, similar to previous plot of replica index.
# change directory
$ cd ../4_plot_potential
$ ls
$ plot_potential.sh plot_potential.gnuplot
# make the file executable and use it
$ chmod u+x plot_potential.sh
$ ./plot_potential.sh
# plot potential energy distribution
gnuplot plot_potential.gnuplot
The file “plot_potential.sh” contains the following commands:
# get potential energies for each temperature
grep "INFO:" ../3_sort/remd_parmID1_trialanine.log | tail -n +2 | awk '{print $2, $5}' > potential_energy_rep1.log
...
...
grep "INFO:" ../3_sort/remd_parmID20_trialanine.log | tail -n +2 | awk '{print $2, $5}' > potential_energy_rep20.log
The file “plot_potential.gnuplot” contains the following commands:
set terminal jpeg
set output "pot-tempretures.jpg"
set key below
set xlabel "Potential Energy kcal/mol"
set ylabel "Probability"
binwidth=50
bin(x,width)=width*floor(x/width)
ndata=2000
plot for [k=1:20] "potential_parmID".k.".log" u (bin($2,binwidth)):(1.0/ndata) t "Tempreture".k with lines smooth freq
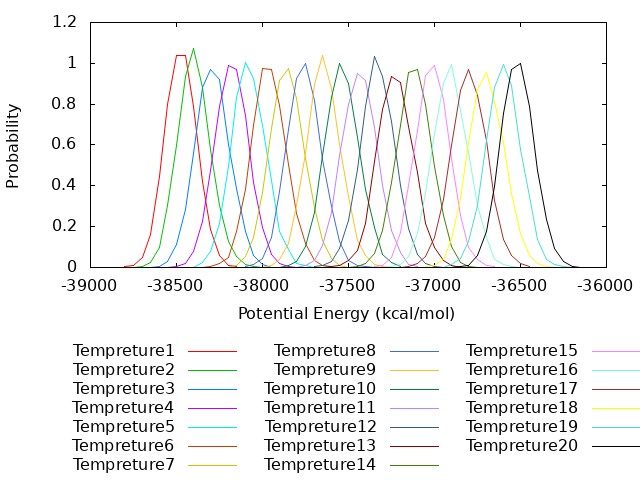
As can bee seen from the figure the potential energies of all temperatures have good overlap.
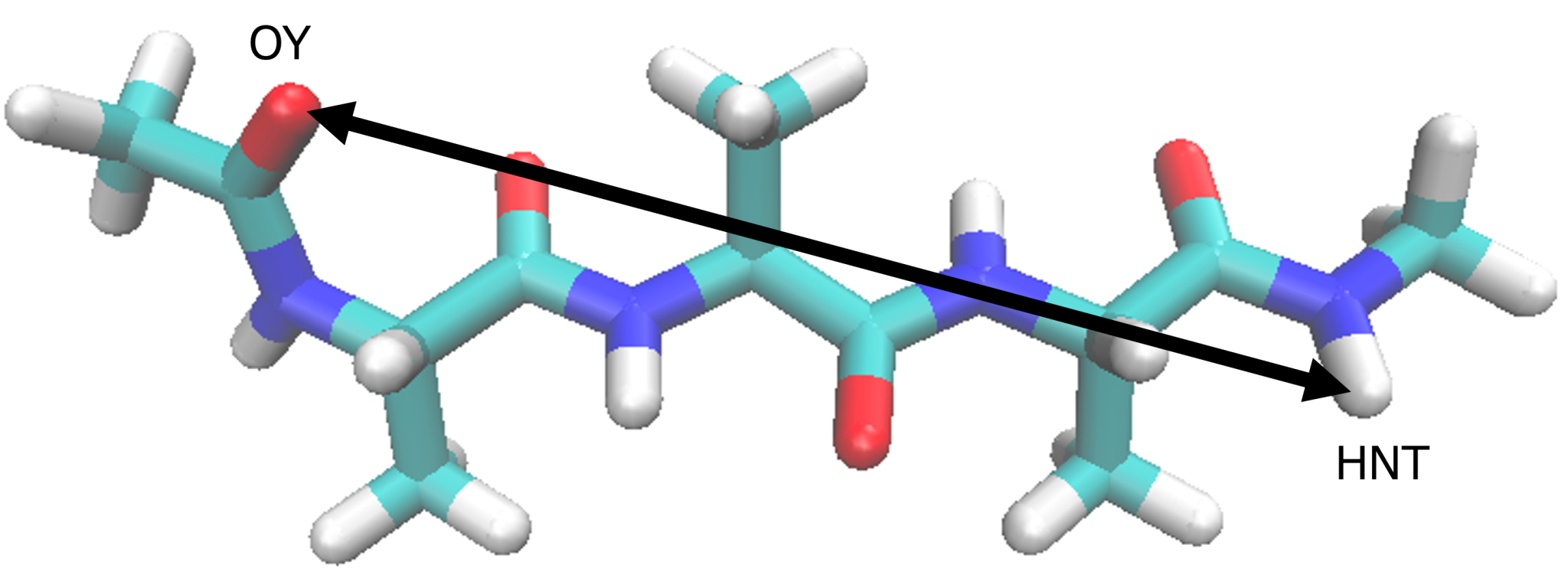
5.5. Calculating end to end distance
In order to calculate PMF (one dmension) of end to end distance
distribution, in the current subsection we calculate the distance
between the two terminal alanine (OY_HNT). In which we use GENESIS
analysis tool trj_analysis as follow:
# change directory
$ cd ../5_end_end_distance
$ ls
$ generate_input_for_trj_analysis.sh run_trj_end_to_end.sh
# make 20 input files
$ chmod u+x generate_input_for_trj_analysis.sh
$ ./generate_input_for_trj_analysis.sh
# run with 20 input files
$ chmod u+x run_trj_end_to_end.sh
$ ./run_trj_end_to_end.sh
One created control file
trj_end_end_parmID1.inp is used to
calculate end to end distance as follow:
[INPUT]
psffile = ../../1_setup/proa.psf
reffile = ../../1_setup/proa.pdb
[OUTPUT]
disfile = parmID1.dis
[TRAJECTORY]
trjfile1 = ../3_sort_dcd/parmID1.dcd
md_step1 = 4000000
mdout_period1 = 2000
ana_period1 = 2000
repeat1 = 1
trj_format = DCD # (PDB/DCD)
trj_type = COOR+BOX # (COOR/COOR+BOX)
trj_natom = 0 # (0:uses reference PDB atom count)
[OPTION]
check_only = NO
distance1 = PROA:1:ALA:OY PROA:3:ALA:HNT
The file run_trj_end_to_end.sh include the following commands:
/home/user/GENESIS/trj_analysis trj_end_end_parmID1.inp | tee trj_end_end_parmID1.log
...
...
/home/user/GENESIS/trj_analysis trj_end_end_parmID20.inp | tee trj_end_end_parmID20.log
5.6. Calculating dihedral angle
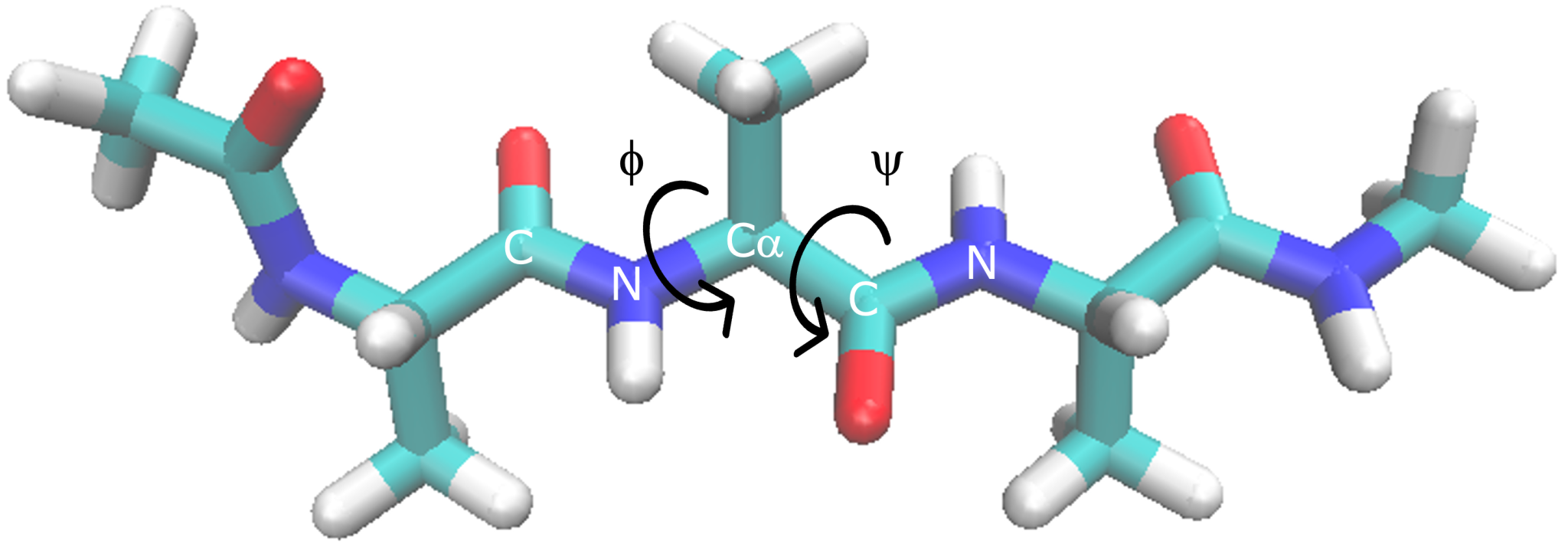
In order to calculate PMF (two dimension) of dihedral angle
distribution, in the current subsection we calculate two dihedral angle
(Φ and Ψ) of 2nd alanine. As with end to end distance, we use GENESIS
analysis tool trj_analysis as follow:
# change directory
$ cd ../6_dihedral_angle
$ ls
$ run_trj_dihedral.sh trj_dihedral.sh
# make 20 input files
$ chmod u+x generate_input_for_trj_analysis.sh
$ ./trj_dihedral.sh
# run with 20 input files
$ chmod u+x run_trj_dihedral.sh
$ ./run_trj_dihedral.sh
One created control file trj_dihedral_parmID1.inp is used to
calculate end to end distance as follow:
[INPUT]
psffile = ../../1_setup/proa.psf
reffile = ../../1_setup/proa.pdb
[OUTPUT]
torfile = parmID1.tor
[TRAJECTORY]
trjfile1 = ../3_sort_dcd/parmID1.dcd
md_step1 = 4000000
mdout_period1 = 2000
ana_period1 = 2000
repeat1 = 1
trj_format = DCD # (PDB/DCD)
trj_type = COOR+BOX # (COOR/COOR+BOX)
trj_natom = 0 # (0:uses reference PDB atom count)
[OPTION]
check_only = NO
torsion1 = PROA:1:ALA:C PROA:2:ALA:N PROA:2:ALA:CA PROA:2:ALA:C
torsion2 = PROA:2:ALA:N PROA:2:ALA:CA PROA:2:ALA:C PROA:3:ALA:N
The file run_trj_dihedral.sh include the followind commands:
/home/user/GENESIS/trj_analysis trj_dihedral_parmID1.inp | tee trj_dihedral_parmID1.log
...
...
/home/user/GENESIS/trj_analysis trj_dihedral_parmID20.inp | tee trj_dihedral_parmID20.log
5.7. MBAR analysis
In order to use conformers from temperatures higher than the target temperature (300 K), we apply GENESIS mbar_analysis tool where we use our sorted potential energy files as cv as follows:
# change directory
$ cd ../7_MBAR
$ ls
$ MBAR.inp
# calculate free energy with MBAR
$ /home/user/GENESIS/bin/mbar_analysis MBAR.inp| tee MBAR.log
The following control file MBAR.inp is used to calculate free
energy:
[INPUT]
cvfile = ../4_plot_potential/potential_parmID{}.log
[OUTPUT]
weightfile = weight{}.dat
fenefile = fene.dat
[MBAR]
num_replicas = 20
input_type = REMD
dimension = 1
temperature = 300.00 302.53 305.09 307.65 310.24 \
312.85 315.47 318.12 320.78 323.46 \
326.16 328.87 331.61 334.37 337.14 \
339.94 342.75 345.59 348.44 351.30
target_temperature = 300.00
tolerance = 10E-08
newton_iteration = 100
self_iteration = 40
This produces fene.dat file containing the evaluated relative free
energies and 20 weight*.dat files containing the weights of each
snapshot for each parameterID. For example, weight1.dat is as follows:
2000 2.792858134883802E-004
4000 1.687935868689458E-004
...
...
3998000 2.803355372323412E-004
4000000 2.787713494107975E-004
5.8. Calculating PMF of distance distribution
The one of the final steps of this tutorial is to use the calculated
distances (5.5). and weight files from MBAR analysis (5.7) to calculate
PMF of end to end distance distribution in alanine tripeptide. We use
another tool in GENESIS pmf_analysis as follow:
# change directory
$ cd ../8_PMF_distance
$ ls
$ PMF.inp plot_pmf.gnuplot
# calculate PMF
$ /home/user/GENESIS/bin/pmf_analysis PMF.inp| tee PMF.log
# plot PMF
$ gnuplot plot_pmf.gnuplot
The following control file PMF.inp is used to calculate PMF about
end to end distance:
[INPUT]
cvfile = ../5_end_end_distance/parameter_ID{}.dis # input cv file
weightfile = ../6_MBAR/weight{}.dat
[OUTPUT]
pmffile = dist.pmf
[OPTION]
nreplica = 20 # number of replicas
dimension = 1 # dimension of cv space
temperature = 300
grids1 = 0 15 61 # (min max num_of_bins)
band_width1 = 0.25 # sigma of gaussian kernel
# should be comparable or smaller than the grid size
# (pmf_analysis creates histogram by accumulating gaussians)
is_periodic1 = NO # periodicity of cv1
The file plot_pmf.gnuplot include the following commands:
set terminal jpeg
set output "PMF_end_end.jpg"
set xrange [0:13]
set yrange [0:10]
set xlabel "End to End Distance (Å)"
set ylabel "PMF (kcal/mol)"
plot "dist.pmf" u 1:2 w l notitle
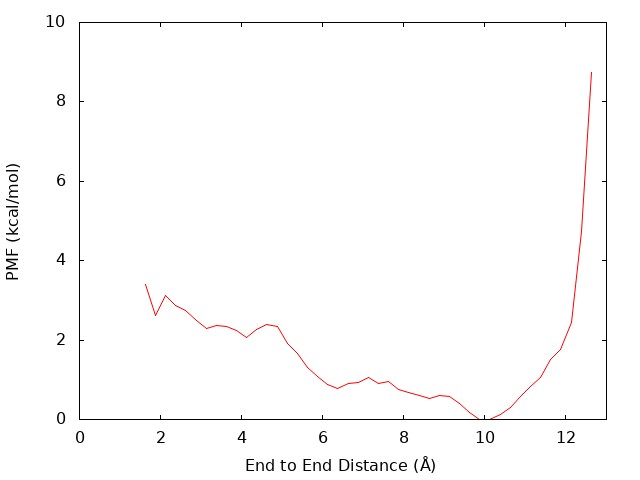
We can see that there is the global energy minimum around r = 10 Å. The latter corresponds to the α-helix conformation, where the hydrogen bond between OY and HNT is formed. These results suggest that in water the (Ala)3 tends to form an extended conformation rather than α-helix.
5.9. Calculating PMF of dihedral distribution
The final step of this tutorial is to use the calculated dihedral angle (5.6) and weight files from MBAR analysis (5.7) to calculate PMF of dihedral distribution of 2nd alanine in (ALA)3.
We can get PMF to the following commands:
# change directory
$ cd ../9_PMF_dihedral
$ ls
$ calc_pmf_2d.py plot_pmf_2d.py
# merge data for python script
cp /dev/null cv.dat
cp /dev/null weight.dat
for i in {1..20}; do
cat ../6_dihedral_angle/parmID${i}.* >> cv.dat
cat ../7_MBAR/weight${i}.dat >> weight.dat
done
# calculate PMF
python3 calc_pmf_2d.py \
-t 300 \
-c 10 \
-n 40000 \
--Xmin -180 \
--Xmax 180 \
--Xdel 10 \
--Ymin -180 \
--Ymax 180 \
--Ydel 10 \
--cv_file cv.dat \
--weight_file weight.dat \
--out_file pmf_2d.dat \
--Xcyclic \
--Ycyclic
# print PMF
python3 plot_pmf_2d.py
Similar to the script of section 6.2 in Tutorial 13.1, the absolute temperature, the number of
samples, the minimum value of the CV, the maximum value of the CV, the
grid size of the bin for the CV are assigned by “-t”, “-n”, “–Xmin”,
“–Xmax”, “–Xdel”, respectively. “-c” represents the cutoff of the
number of samples in each bin. If the number of samples in a certain bin
is below 10, the probability (or free energy) in the bin is omitted.
“–Xcyclic” represents the periodicity of the CV. This script outputs
the free energies in each bin “pmf.dat” and grid points of the CVs
“xi.dat” and “yi.dat”. plot_pmf_2d.py draws the two-dimensional
free-energy landscape using “pmf.dat”, “xi.dat”, and “yi.dat”.
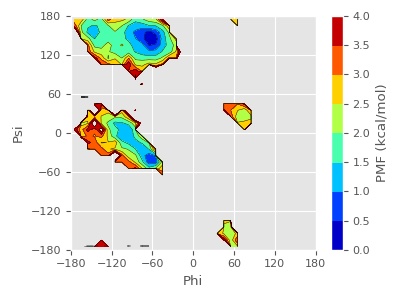
We can see that there is the global energy minimum around (φ,ψ) = (-75°, 150°) and local energy minimum around (φ,ψ) = (-60°, -45°). The latter corresponds to the PPII and α-helix conformation respectively. As in the PMF about end to end distance in (5.7), we can understand that (Ala)3 in explicit water prefer extended conformation rather than α-helix.
References
Written by Daisuke Matsuoka@RIKEN Theoretical molecular science
laboratory
Updated by Hisham Dokainish@RIKEN Theoretical molecular science
laboratory
August, 28, 2019
Updated by Daiki Matsubara@RIKEN Center for Biosystems Dynamics Research
(BDR)
March, 31, 2022