GENESIS Input Example: Hydrogen bonds (hbond_analysis)
hbond_analysis is a member of
SPANA and the parallelized
version of
hb_analysis.
This tool is parallelized with hybrid MPI/OpenMP. Like hb_analysis,
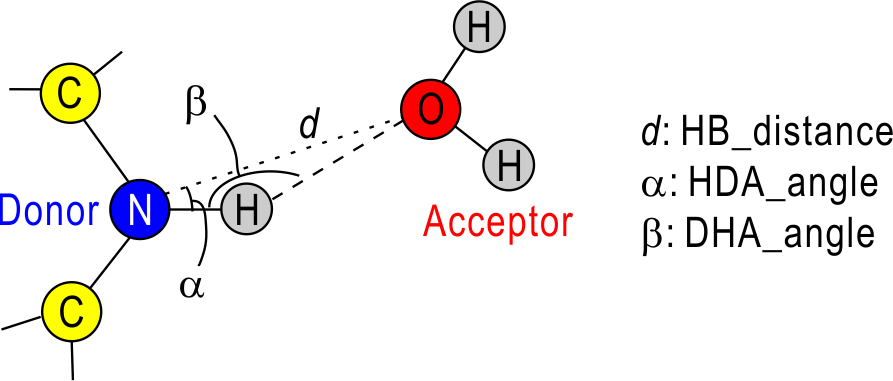
we need the three geometrical parameters for the detection of H-bond:
hb_distance (d), HDA_angle (α), and DHA_angle (β). In the
default, d = 3.4 Å, α = 30.0 deg. and β = 120.0 deg, and if you use
your own definition, you can specify these value in [HBOND_OPTION].
Hbond_analysis tool is useful to examine H-bond patterns for a huge protein
molecule or for crowding system. You can limit the range used for the analysis
(for example, focus on H-bonds around ligand-binding site or in hydrophilic
channel) by adequately using determine_box = MANUAL option.
Functions of this tool is same as hb_analysis except this analysis is done
with space_decomposition. Thus, when you need a H-bond list written in
hb_listfile, the hb_listfile is output per domain. When you specify
hb_listfile name, you need () to return MPI process numbers. The parameter
cutoff is important to determine the range to search H-bond partners of
analysis_atom. The cutoff value should be larger than the hb_distance
value.
Unlike hb_analysis, to wrap molecules into the PBC box, we need the use of the
options recenter and wrap to correctly detect H-bonds formed between
analysis_atom and target_atom.
[INPUT]
psffile = BPTI_ionize.psf
reffile = BPTI_ionize.pdb
[OUTPUT]
txtfile = BPTI-wat_Hbond.txt
hblist_file = BPTI_wat_hbond_mpi().list
[TRAJECTORY]
trjfile1 = run.dcd
md_step1 = 10
mdout_period1 = 1
ana_period1 = 1
trj_format = DCD
trj_type = COOR+BOX
[ENSEMBLE]
ensemble = NPT
[BOUNDARY]
type = PBC
domain_x = 2
domain_y = 2
domain_z = 1
num_cells_x = 10
num_cells_y = 10
num_cells_z = 10
box_size_x = 50.0
box_size_y = 50.0
box_size_z = 50.0
[SELECTION]
group1 = sid:BPTI & resno:1-58
group2 = resname:TIP3
group3 = sid:BPTI & (resno:3 | resno:7-8 | resno:39-40 | resno:43)
[SPANA_OPTION]
wrap = yes # wrap molecules if wrap = yes
buffer = 5.0 # this distance should be larger than hb_distance.
box_size = TRAJECTORY # (TRAJECTORY / MANUAL / MAX)
[HBOND_OPTION]
recenter = 3
output_type = count_atom # (count_atom / count_snap)
analysis_atom = 1
target_atom = 2
solvent_list = TIP3
# hb_distance = 3.4 # upper limit of distance between Donor and Acceptor atoms, default value is 3.4 A
# dha_angle = 30.0 # upper limit of H-bond D-H..A angle, default value is 30.0 deg.
# hda_angle = 120.0 # lower limit of H-bond H-D..A angle, default value is 120.0 deg.