General Introduction of SPANA
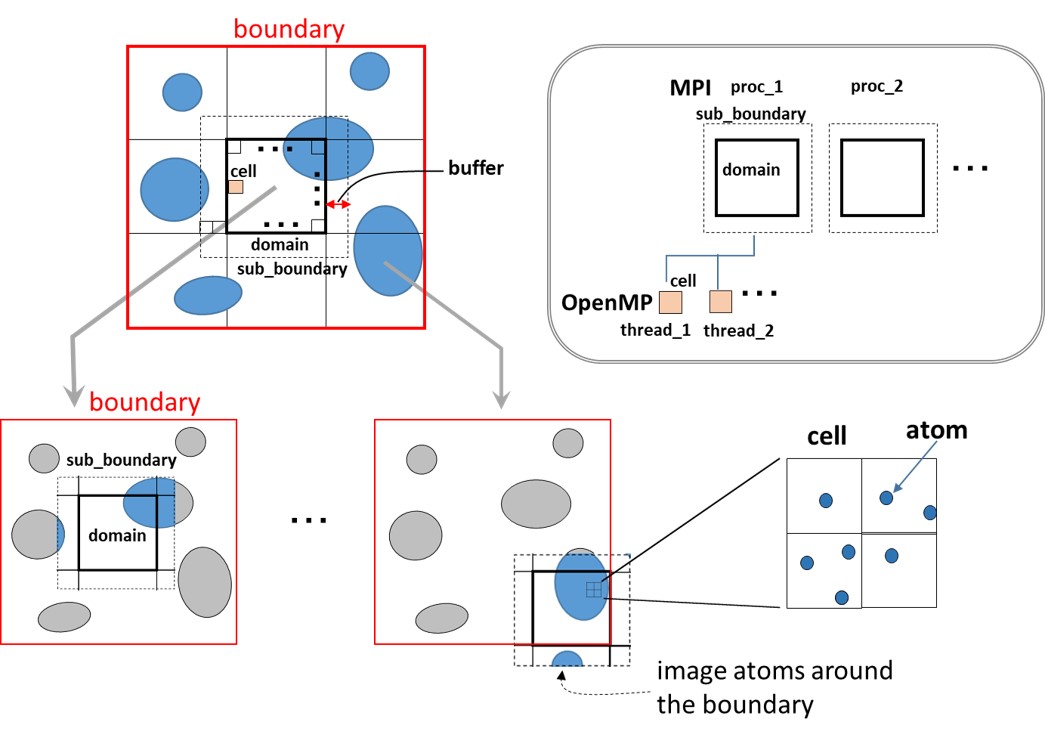
SPANA adopts MPI/openMP hybrid parallelization scheme. In this scheme, a
boundary is set for each snapshot structure you want to analyze. Generally, the
dimension of this boundary is equal to that of the Periodic Boundary Condition
(PBC) box. In the MPI parallelization, the boundary is decomposed into smaller
domains based on parameters domain_[x, y, z] specified in [BOUNDARY]
section. Each domain is further decomposed into cells with parameters
num_cell_[x, y ,z] in [BOUNDARY] section. Each domain has buffer region with
thickness specified by a parameter “buffer” in [SPANA_OPTION] section. Here,
we call the domain+buffer region ‘sub_boundary’. Each MPI process shares the
same molecule information defined in a psf file or a prmtop file and in a PDB
file. However, each MPI process selectively assigns atoms located inside the
sub_boundary into cells. This approach reduces memory usage required for cell
information, and enables each MPI process to get the atom information (such as
serial index and xyz coordinates) in target cells in the sub_boundary.
Calculation for each domains is done by each MPI process, and calculation for
cells are decomposed into OpenMP threads.
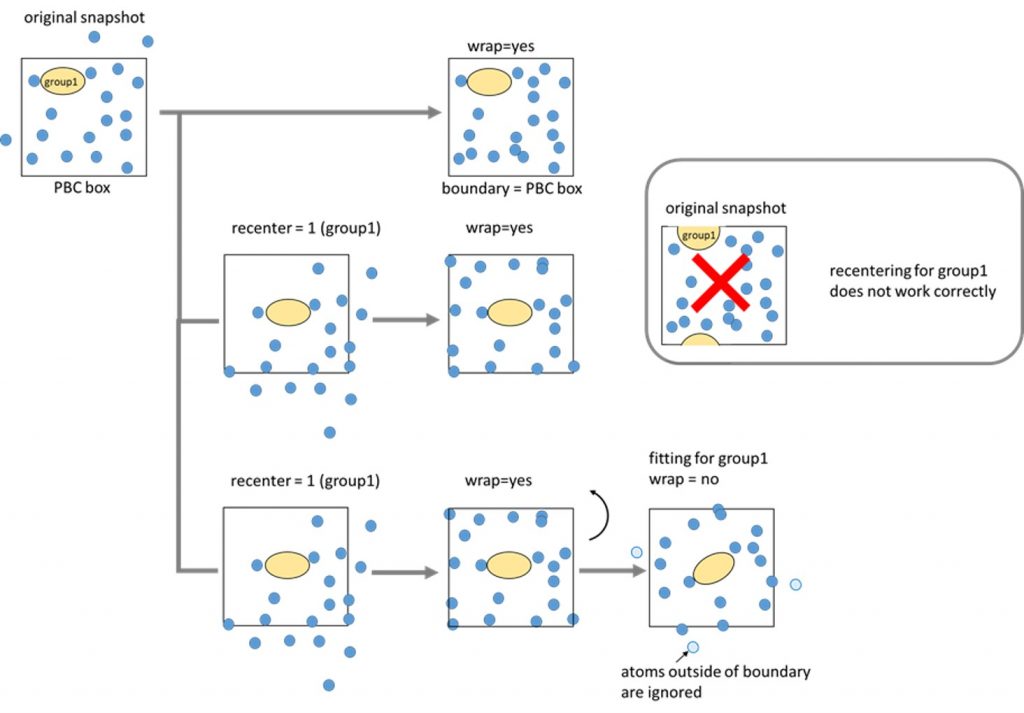
When [BOUNDARY] type = PBC and [SPANA_OPTION] wrap = yes, all atoms are
wrapped into the unit PBC box using the box size information written in the
trajectory file (dcd file). If the file does not have the box size information,
the option wrap = yes does not work. When wrap = no, the program does
nothing for atomic coordination. Some SPANA application (e.g. rdf_analysis)
has an option recenter. The vector from the center of the unit box to the
center of mass of the specified group is calculated, and all atoms are
translated using this vector. In this case, wrapping of the atomic coordinates
is performed after the centering. When you use the option recenter, please
make sure that the target solute molecule is not chopped with PBC boundary. If
recenter = 0, or the recenter option does not appear in a control file, the
centering is not done. Some SPANA application (e.g., density_analysis) uses
fitting options in [FITTING] section. In this case, it is recommended to
recenter the group for fitting. The fitting will be performed after the
recentering and wrap. Note that after the fitting, atoms outside of the boundary
are not wrapped again, and are ignored by analysis.
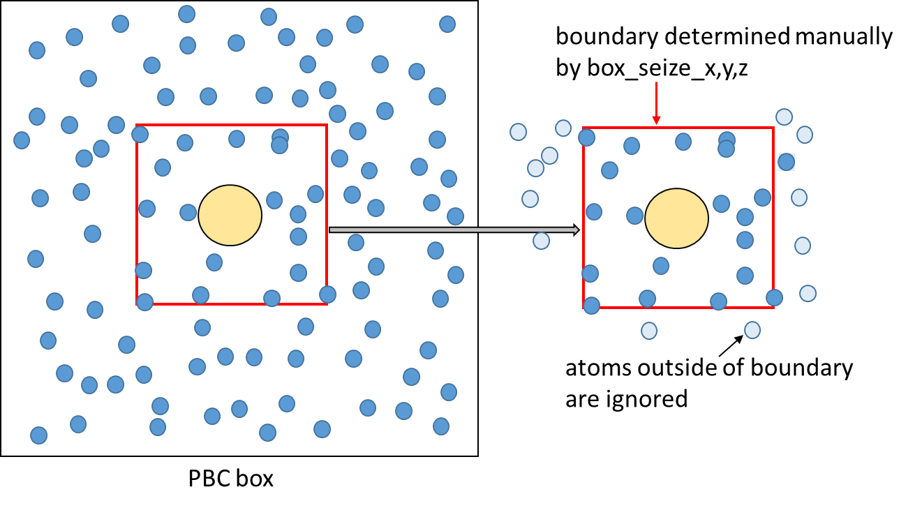
The boundary of the whole system is determined by option box_size in
[SPANA_OPTION] section. In the case of [BOUNDARY] type = PBC and box_size =
trajectory, the boundary is determined from the box information written in the
trajectory file. Therefore, the boundary size is the same as that of PBC box. On
the other hand, when box_size = manual, the boundary size is determined from the
values box_size_[x, y, z] in [BOUNDARY] section. This option is useful to
save the memory usage, because, when this option is used, the program performs a
calculation only for the selected region around the target solute molecule. In
this case, atoms outside of the manually determined boundary are not assigned
into any cells and ignored by analysis. Please note that the target molecule
should be centered when you use box_size = manual.
When [BOUNDARY] type = NOBC, the boundary used for the analysis should be
determined with box_size = manual.
General example of SPANA input file
The following sample is the typical input for the SPANA. This input is used to
calculate the radial distribution function of hydration water (see
rdf_analysis for detail) around BPTI (group1) in water box with PBC. The
whole system is decomposed into 8 domains by domain_[x, y, z] = 2.
Furthermore, the whole system is decomposed into 136 x 160 x 132 cells by
num_cell_[x, y, z]. The boundary size is determined by PBC box size written in
the trajectory file. In every frame for the analysis, coordinates of all atoms
are translated by the vector used for the centering of BPTI (recenter = 1).
After the translation, all atoms are wrapped (wrap=yes) inside the PBC box.
The thickness of the buffer region (i.e., thickness of the sub_boundary around
domain) is set to 10 by the option cutoff.
[INPUT]
psffile = ionize.psf
reffile = ionize.pdb
pdbfile = ionize.pdb
[OUTPUT]
txtfile = bpti_proximal.out
[TRAJECTORY]
trjfile1 = run.dcd
md_step1 = 500000
mdout_period1 = 500
ana_period1 = 5000
repeat1 = 1
trj_format = DCD # (PDB/DCD)
trj_type = COOR+BOX # (COOR/COOR+BOX)
[BOUNDARY]
type = PBC
box_size_x = 68.25815
box_size_y = 80.24045
box_size_z = 66.58892
domain_x = 2
domain_y = 2
domain_z = 2
num_cells_x = 136
num_cells_y = 160
num_cells_z = 132
[SELECTION]
group1 = ai:1-892
group2 = rnam:TIP3 and an:OH2
[SPANA_OPTION]
buffer = 10
wrap = yes
box_size = TRAJECTORY
[RDF_OPTION]
rmode = proximal
solute = 1
solvent = 2
binsize = 0.25
range = 10
bulk_region = 15.0
recenter = 1